Pharmacokinetic modeling for long-acting (LA) medicines
Speaker: Andrew Owen, Professor of Molecular and Clinical Pharmacology at University of Liverpool

Demonstrated how modeling can be used in the development of long-acting (LA) medicines using three projects from the LEAP Modeling and Simulations Core.
Modeling can help across development of new long-acting (LA) medicines.
- Role of physiologically based pharmacokinetic (PBPK) modeling – understand drug compatibility with LA delivery using early in-vitro drug disposition data; inform dose selection for animal studies (in vitro-in vivo extrapolation) and Phase 1 trials; and guide clinical management and optimization (e.g. pregnancy/lactation, neonates/pediatrics, drug-drug interactions, dose optimization and genetics).
- Role of other pharmacometrics approaches – shed light on unexplored mechanisms (alongside animal data); assess exposure-response relationships throughout clinical development (efficacy and safety); and guide clinical management/optimization (with PBPK modeling).
- Confidence in model outcomes is generally proportional to the quality of input data and increases as medicines progress through the phases of development and understanding of the medicines grows.
LEAP Example 1: PBPK modeling to inform development – CAB microarray patch (MAP) in rats and humans.
- Base model of MAP for LA ARVs (published several years ago).
- Unqualified model describes the microneedles (MN) and drug release from various parts of the MN into the striatum corneum, viable epidermis and dermis.
- Looked at drug penetration and partition coefficient from each site into the blood and lymph compartment of a simulated population.
- Model qualification (simulated formulation performance vs empirical PK data) against pre-clinical CAB MAP and clinical CAB LAI PK data.
- Rat single-dose CAB MAP qualification, Rat multiple-dose CAB MAP qualification and human LAI CAB qualification – good fit between empirical data and simulated CAB performance generates confidence in the modeling.
- Predicted CAB MAP performance in humans.
- Simulated different CAB doses (75mg, 150mg and 300mg) delivered through a MAP (unpublished) to generate expected PK performance in humans based on everything that has been qualified about the model.
LEAP Example 2: PBPK modeling to inform deployment – dose prediction for LAI CAB in neonates.
- Model qualification against adult clinical LAI CAB PK data.
- Adjusted model parameters with known differences in neonatal populations – recognizing that residual uncertainty exists, can have reasonable confidence in the model (i.e. mechanisms described by the model and formulation performance through the model) to simulate LAI CAB performance in neonates.
- Simulated neonatal exposures across different CAB IM doses assuming adult release kinetics.
- Regimen 1 (CAB 20mg IM): target plasma CAB concentrations (above 4xPAIC90) are achievable, but neonatal simulation unveiled a delay in CAB absorption, not seen in adults.
- Regimen 2 (CAB 20mg IM + CAB 0.8mg oral): target plasma CAB concentrations can be achieved within the first day by adding a single, oral CAB dose at Day 0.
LEAP Example 3: Different pharmacometrics approaches to rationalize mechanisms – a Centre of Excellence in Long-acting Therapeutics (CELT) LAI development project.
- Mechanistic knowledge underlies PBPK modeling, yet fundamental understanding is limited for many LA technologies, particularly regarding drug absorption processes.
- Simple model to fit empirical animal PK data for the active pharmaceutical ingredient (API) (exposure up to 28 days) showed dose linearity across 50, 100 and 200 mg/kg doses of the formulation.
- Pharmacometrician de-convoluted available IV and IM PK data to derive an in vivo release profile for each dose – predicted release kinetics unveiled bi-exponential release, supporting a parallel fast and slow input from the depot into the systemic circulation.
- Constructed a more advanced model describing the drug depot with direct release into the systemic compartment (early, fast release) and a 2-compartment transit model into the systemic compartment (longer duration, slow release)
- Modeling cannot elucidate the mechanisms, but does fit with rapid absorption of the soluble component of the formulation and slower release of the solid and achieves a much better fit to the empirical PK data.
LEAP Modelling and Simulation Core is a resource for the LA research community –
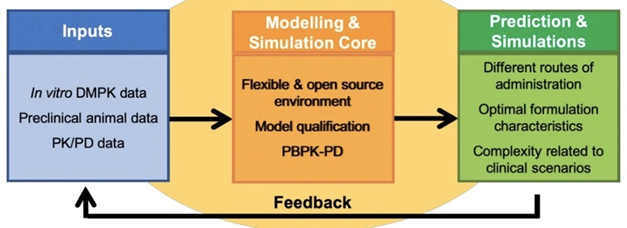
- Our template submission form details the input data needed for us to engage with you. https://logactinghiv.org/files/Modeling-Core-Submission-Form.docx